Translation: Français, Русский, 中文
What is Mendelian randomization? – a brief guide:
This guide is intended for newcomers to the field. It is written to be accessible to academics, journalists, and interested members of the public, so please forgive any technical inexactness – we have tried to simplify the explanations as far as possible without introducing any inaccuracies. If you aren't sure what a word means, try hovering over it - you may find a definition or an example!
Mendelian randomization uses genetic variants to make a judgement about the causal nature of the relationship between a risk factor and an outcome based on observational data. Taking that one step at a time…
The aim – understanding causal relationships: We want to learn about the status of causal relationships between proposed risk factors and disease outcomes so we can answer important questions like: “Is alcohol consumption a causal risk factor for liver cancer?”. The ideal way to do this is a randomized trial, in which we randomize some individuals to an intervention on the risk factor (the treatment group) and some to no intervention (the control group). We then wait and compare levels of the outcome between the groups.
The challenge – obtaining randomized evidence: However, this approach faces many challenges in practice. Randomized trials are long and expensive, and in many cases impractical or unethical. Could we establish a randomized trial to test the effect of alcohol consumption on liver cancer risk? Would you be willing to volunteer to take part in a study where the toss of a coin determined whether you drink alcohol or not for the next 30 years? Hence, we often need to learn about causal relationships from observational studies where there is no randomized intervention.
The problem – correlation without causation: Observational studies are influenced by reverse causation and confounding. Individuals with poor health may choose to cut down on their alcohol consumption (an example of reverse causation). Reverse causation can lead to an association between risk factor and disease, but it would be disease causing changes in the risk factor, not the other way round. In this case, reverse causation could hide the true causal relationship, as those labelled as 'non-drinkers' would include lifelong abstainers and recent quitters - thus reducing the true difference in alcohol consumption between drinkers and non-drinkers.
Alcohol drinkers and non-drinkers differ in many ways other than alcohol consumption – for example, alcohol drinkers tend to be more likely to smoke, which may affect liver cancer risk (an example of confounding). While we can adjust for these factors in a statistical analysis, we can never know if we have accounted for all of them. Both reverse causation and confounding can lead to risk factors and outcomes being associated, but without a causal relationship – hence the saying: “correlation is not causation”.
As a result of these biases, many reported associations from observational studies do not have a causal basis. This leads to confusing and often contradictory health messages, and leaves people wondering – what actually is good for me?
The solution? – Mendelian randomization: Mendelian randomization is a statistical approach with the potential to avoid such biases. The technique assesses whether genetically-predicted levels of a risk factor and a disease outcome are associated or not (or equivalently, whether genetic variants that predict levels of the risk factor are associated with the outcome or not). As Mendel’s law of independent assortment states that characteristics are inherited independently of each other, genetic associations are less susceptible to confounding. Furthermore, as our genetic code is established before birth, there is less potential for reverse causation – the genetic variants must be at the start of any causal chain of events. Therefore, associations in a Mendelian randomization study are more likely to have a causal interpretation than those from conventional epidemiological analyses.
In short, if specifically-chosen genetic variants that influence levels of the risk factor are associated with the outcome, this provides evidence that the risk factor has a causal effect on the outcome.
Resources:
Two-minute video introduction to Mendelian randomization: https://www.youtube.com/watch?v=LoTgfGotaQ4
A longer introduction aimed at an informed, but lay audience: https://phw.nhs.wales/publications/publications1/making-sense-of-mendelian-randomisation-and-its-use-in-health-research/
Introductory lecture by George Davey Smith (~40 minutes): https://www.youtube.com/watch?v=Whut4Yo-x-A
Deeper explanation - worked example of C-reactive protein and heart disease risk:
We can understand better how Mendelian randomization works in practice by considering an example. “C-reactive protein” is a marker of inflammation that has been proposed as a potential causal risk factor for several diseases. It is observationally associated with heart disease risk. But does this association reflect a causal relationship? Is C-reactive protein really a causal risk factor for heart disease?
|
Genetic variants in the CRP gene region affect C-reactive protein levels – in fact, that is why the gene region is called the CRP gene region. It is biologically plausible that these genetic variants affect C-reactive protein levels in a specific way. That is, they change the levels of C-reactive protein, but do not affect other risk factors. Indeed, empirically we can see that these variants are not associated with potential confounders: body mass index, blood pressure, cholesterol, and so on (see Figure 1). Let’s simplify and assume the genetic variants divide the population into two groups, which we call genotype A and genotype B. We also assume that people with genotype A have higher average levels of C-reactive protein, and people with genotype B have lower average levels of C-reactive protein. But, due to the nature of genetic inheritance, the two groups don’t have different average levels of body mass index, blood pressure, cholesterol, and so on. Under our assumptions, any association between the genotype groups and heart disease risk can only occur if C-reactive protein is a causal risk factor. |
 |
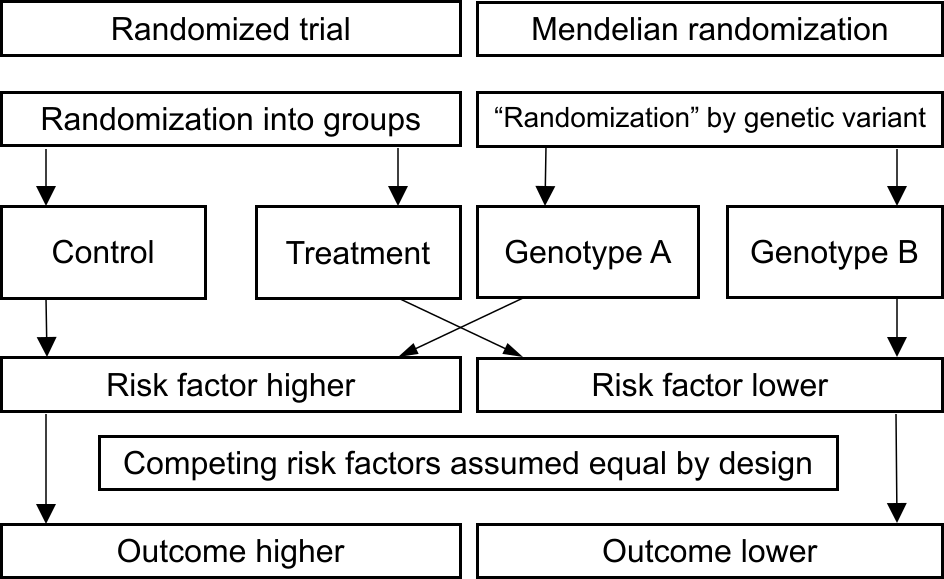 |
Here, the presence or absence of the genetic variant is similar to random assignment in a randomized trial (see Figure 2). In a randomized trial, we would assess the impact of an intervention on C-reactive protein levels. For example, by introducing a pill that lowers C-reactive protein and randomly assigning individuals either to take this pill or to take a placebo. We would then compare outcomes between those assigned to take the pill versus those assigned to take the placebo. In Mendelian randomization, we compare those with genotype A versus those with genotype B. If risk of heart disease differs between these genetically-defined groups, then we would interpret this as evidence that if we changed C-reactive protein levels, that would affect heart disease risk - in other words, this would provide evidence that C-reactive protein is a causal risk factor. |
When we performed this analysis in reality (https://www.bmj.com/content/342/bmj.d548), we observed no association between the genotype group and heart disease risk (or equivalently, no association between genetically-predicted levels of C-reactive protein and heart disease risk) for four genetic variants in the CRP gene region. Hence, using Mendelian randomization we found no evidential support for C-reactive protein as a causal risk factor for coronary heart disease.
Some questions:
Can Mendelian randomization prove that a risk factor is a cause of disease risk?:
In short, no. All approaches that make causal judgements based on observational data rely on untestable assumptions. In the case of Mendelian randomization, the untestable assumptions are that the genetic variant(s) are distributed independently of potential confounders, and the genetic variant(s) can only affect the outcome via the risk factor. Technically speaking, these assumptions mean that we are treating the genetic variant(s) as instrumental variables.
Mendelian randomization can provide supportive evidence for a causal relationship from observational data, but it cannot prove or demonstrate a causal effect. We are trying to ensure that researchers use appropriate language with respect to causal claims (see https://jamanetwork.com/journals/jamacardiology/article-abstract/2770709) – sometimes they do, other times they are not as careful as they should be!
How reliable are claims from Mendelian randomization?:
Some are more reliable, others are less reliable. In the example above, the genetic variants considered were in the CRP gene region – the part of the genetic code that tells the body how to make C-reactive protein. So it is plausible to argue that these genetic variants influence C-reactive protein in a specific way, and hence the only potential causal pathway from these genetic variants to heart disease risk runs via C-reactive protein levels.
Similarly, there are genetic variants in the HMGCR gene region that influence LDL-cholesterol (so called “bad cholesterol”) in a specific way – and in doing so, they mimic the action of statins, a widely prescribed class of drugs. Statins reduce the risk of coronary heart disease and ischaemic stroke, and similarly, genetic variants in the HMGCR gene region are associated with coronary heart disease and ischaemic stroke risk. However, while statins overall increase longevity, they do slightly increase risk of Type 2 diabetes; similarly, these genetic variants are associated with Type 2 diabetes risk, and the association is in the opposite direction to that with heart disease.
There are several examples where Mendelian randomization results and randomized trial results tell the same story, and increasingly, examples where the Mendelian randomization analysis has predicted the outcome of the randomized trial (for example, the use of tocilizumab and baricitinib as treatments for COVID-19).
However, other examples are less clear. For example, if the risk factor is length of sleep duration – is it really plausible that genetic variants affect sleep duration in a specific way? There is no section of the genetic code that directly tells the body how long to sleep for. Hence a Mendelian randomization analysis that shows an association between genetically-predicted sleep duration and lower cancer risk might be useful in providing some evidence that sleep is a causal risk factor for cancer. However, it would be unwise to claim that the investigation is definitive. The analysis can provide some evidence on an important research question, but the Mendelian randomization evidence alone is not conclusive.
How can you tell the difference between a reliable Mendelian randomization investigation and a less reliable one?:
This is a difficult question of judgement, and skilled researchers may come to different conclusions about the reliability of a particular investigation. There are many factors to take into account, but the critical questions are:
- To what extent do the genetic variants mimic an intervention on the risk factor?
- How consistent is the evidence that genetic predictors of the risk factor in different gene regions are associated with the outcome?
For the first question, genetic variants are more likely to reflect an intervention on a molecular risk factor, such as C-reactive protein or cholesterol levels, particularly when the function of the gene is known to relate to the risk factor. Genetic variants are less likely to reflect an intervention on a social or behavioural risk factor, such as number of years spent in education or length of sleep duration. This is because there are many factors that influence these risk factors, so there are many possible causal pathways from the genetic variants to the disease outcome – some of these may not pass through the risk factor.
For the second question, researchers should provide enough information to assess this point. If researchers show that 10 genetic predictors of the risk factor in 10 different gene regions are all associated with the outcome, and if all the associations are in the same direction (i.e. they all suggest that the risk factor is harmful, or all suggest it is protective), then it is plausible that the risk factor has a causal effect on the outcome. However, if only one out of 10 genetic predictors of the risk factor is associated with the outcome, then there may be a causal pathway from that variant to the outcome not via the risk factor – this is known as pleiotropy.
Are genetic variants really randomized?:
Genetic variants are not strictly randomized for all individuals in a population (it is only possible to inherit a genetic variant if one of your parents had it). However, there is some randomization in terms of which sperm cell met which egg cell, as well as how your parents met, which means that most common genetic variants can be treated as being randomly distributed in the population. In other words, we can reasonably assume they are distributed independently of confounding variables – this is the crux of what we mean by “randomized”. Additionally, genetic variants that are not physically closely located on the chromosome tend to be inherited independently; this is Mendel’s law of independent assortment. Technically speaking, we call this quasi-randomization, not true randomization.
There are lots of caveats to this statement. For example, some genetic variants are more common in certain ethnic groups. If a genetic variant is more common in a particular ethnic group, then it would be associated with factors that are more prevalent in that group. Nevertheless, there is lots of empirical evidence to suggest that this assumption is a reasonable starting point in well-mixed population groups (see https://pubmed.ncbi.nlm.nih.gov/18076282/ and https://www.biorxiv.org/content/10.1101/124362v1).
How can I learn more?
Here are some links to key papers that go deeper into explaining the Mendelian randomization paradigm.
Overview of Mendelian randomization: https://www.annualreviews.org/doi/abs/10.1146/annurev-genom-083117-021731
Guidelines for performing Mendelian randomization investigations: https://wellcomeopenresearch.org/articles/4-186/v3
Guidelines for reporting Mendelian randomization investigations: https://www.bmj.com/content/375/bmj.n2233
TL;DR (100 word version): Observational epidemiological studies are influenced by reverse causation and confounding. Mendelian randomization is an epidemiological approach with the potential to avoid such biases. The technique assesses whether genetically-predicted levels of a risk factor (such as coffee drinking) and a disease outcome (such as cancer) are associated. By Mendel’s laws, characteristics are inherited independently of each other, meaning genetic associations are less susceptible to confounding. Furthermore, as genetic variants are established from birth, the potential for reverse causation is diminished. Therefore, associations in a Mendelian randomization study are more likely to have a causal interpretation than those from conventional epidemiological analyses.
If that isn’t enough, please buy the Mendelian randomization book or attend one of our courses: our research group runs an online course on Mendelian randomization twice yearly – we would love for you to attend!