- Details
- Written by: Steve Burgess
The MendelianRandomization software package enables the implementation of various methods for Mendelian randomization using summarized data. This package has recently been updated a number of times to add extra functionality, and is now on version 0.9.0.
Some references:
- MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. This describes the basic operation of the package and core functions.
- MendelianRandomization v0.5.0: updates to an R package for performing Mendelian randomization analyses using summarized data. This describes updates to the package up to version 0.5.0.
- MendelianRandomization v0.9.0: updates to an R package for performing Mendelian randomization analyses using summarized data. This describes updates to the package up to version 0.9.0.
Two other brief updates to announce:
First, we have released MRZero, a slimmed-down version of the Mendelian randomization package. This contains 97% of the same functionality of the original MendelianRandomization package, but with fewer dependences (including no C++ content), and less extraneous content (no exemplar PhenoScanner dataset and no vignette). Overall, while MendelianRandomization is ~1000 kB, MRZero is only 106 kB. This version of the package is designed for resource-limited settings, and for quick installation on a new computer when you don’t want to load so many dependent packages.
Second, we have updated the vignette of the MendelianRandomization package to version 0.9.0. This will be distributed together with the next version of the package, but until then it is available for download here, or available in interactive form here.
We hope you enjoy the new functionality in the package, and we look forward to further suggestions on how to make the package better!
- Details
- Written by: Steve Burgess
Non-linear Mendelian randomization (or stratified Mendelian randomization) is an extension to standard Mendelian randomization that performs separate instrumental variable analyses in strata of the population. The motivation is to understand the causal dose—response curve relating the exposure to the outcome – how does the outcome vary on average when we intervene on the exposure at different levels of the exposure?
The initial proposal for this method was to stratify the population on the “residual exposure” – that is, residual values from regression of the exposure on the genetic variants. This can be performed fairly simply in R, but here are some coding shortcuts. We first load the SUMnlmr package, and use this to generate synthetic data:
devtools::install_github("amymariemason/SUMnlmr")
library(SUMnlmr)
set.seed(31415)
test_data<-create_ind_data(N=10000, beta2=2, beta1=1)
outcome = test_data$quadratic.Y
exposure = test_data$X
grs = test_data$g
We calculate the residual exposure by regressing the exposure on the genetic score, subtracting the fitted value from the measured value, and then adding the overall mean (to ensure that the values of the residual exposure are interpretable):
exposure0 = exposure - lm(exposure ~grs)$fit + mean(exposure)
We can divide into a fixed number of equally-sized quantiles:
quant = 10
qs = quantile(exposure0, prob=seq(0, 1-1/quant, by=1/quant))
quantx0 = as.numeric(lapply(exposure0, function(x) { return(sum(x>=qs)) }))
Or into strata at fixed values of the residual exposure (here <0.5, 0.5-1.0, 1.0-1.5, 1.5-2.0, and >2.0):
quantx0 = as.numeric(cut(exposure0, breaks=c(-Inf,0.5,1,1.5,2,Inf), include.lowest=TRUE))
And then calculate genetic associations with the exposure and outcome within each stratum:
bx=NULL; bxse=NULL; by=NULL; byse=NULL; xmean=NULL
for (j in 1:length(unique(quantx0))) {
bx[j] = lm(exposure[quantx0==j] ~ grs[quantx0==j])$coef[2]
bxse[j] = summary(lm(exposure[quantx0==j] ~ grs[quantx0==j]))$coef[2,2]
by[j]= summary(lm(outcome[quantx0==j] ~ grs[quantx0==j]))$coef[2,1]
byse[j] = summary(lm(outcome[quantx0==j] ~ grs[quantx0==j]))$coef[2,2]
xmean[j] = mean(exposure[quantx0==j])
} # add covariates to the regression models to taste
Alternatively, we can use the built-in function create_nlmr_summary, which does this for equally-sized quantiles in one step:
summ_data <-create_nlmr_summary(y = outcome,
x = exposure,
g = grs,
covar = NULL, # add covariates here
family = "gaussian",
# gaussian for continuous outcome, binomial for binary outcome
strata_method = "residual",
controlsonly = FALSE,
# only relevant for binary outcomes
q = 10) # number of quantiles
Once we have the summarized data, we could calculate IV estimates for each stratum and stop there. Alternatively, we could use one of the fancy plotting functions in the SUMnlmr package to plot the data:
frac_poly_summ_mr(bx=bx,
by=by,
bxse=bxse,
byse=byse,
xmean=xmean,
family="gaussian",
fig=TRUE)
Similarly, there is the piecewise_summ_mr function. There are lots of options in both functions here to explore.
One point to clarify is that the IV estimates represent the change in the outcome per unit difference in the genetically-predicted values of the exposure (or with a causal interpretation, the change in the outcome per unit increase in the exposure). Whereas the plotting functions give the estimated relationship between the exposure and outcome. In these plots, the IV estimate is the gradient (slope) of the curve. So these options represent two ways of expressing the same information.
An assumption made in the “residual method” for non-linear Mendelian randomization is that the genetic effect on the exposure is constant in the population. It turns out that this assumption is often violated. We have recently developed a non-parametric method (the “doubly-ranked method”) for stratifying the population that is less sensitive to violations of this “constant genetic effect” assumption. This can be implemented in the SUMnlmr package using the strata_method = "ranked" option:
summ_data2<-create_nlmr_summary(y = outcome,
x = exposure,
g = grs,
covar = NULL,
family = "gaussian",
strata_method = "ranked",
controlsonly = FALSE,
q = 10)
Our current advice is to assess the constant genetic effect assumption using the doubly-ranked method – are the bx estimates from the doubly-ranked method similar? (It turns out that the bx estimates from the residual method are similar whether this assumption is satisfied or not – something we didn’t initially realise.)
If the assumption is violated, then: 1) compare non-linear MR results from both methods (residual and doubly-ranked), with more weight on results from the doubly-ranked method, as this method is less sensitive to violations of the assumption; and 2) consider transforming the exposure – if the bx estimates are increasing, then a log transformation may be appropriate. Transformation will not change the by estimates from the doubly-ranked method, and so transformation will not affect reliability of the doubly-ranked method, but it will change the bx estimates – are these now more similar? If so, results from the residual method following transformation will be more reliable. We note that transformation of the exposure does change the interpretation of the bx estimates and consequently the interpretation of the IV estimates.
> summ_data2
$summary
bx by bxse byse xmean xmin xmax
1 0.2679257 3.057584 0.01066202 0.1345234 2.569048 2.327217 2.957322
2 0.2554275 3.174083 0.01166755 0.1602636 2.801911 2.437782 3.187628
3 0.2602369 3.532592 0.01243878 0.1748998 2.980195 2.605649 3.382229
4 0.2521341 3.577799 0.01326634 0.1946739 3.149608 2.766214 3.554172
5 0.2471285 3.615373 0.01482321 0.2272206 3.336171 2.932134 3.804060
6 0.2442914 3.703393 0.01775254 0.2868735 3.537327 3.070300 4.031026
7 0.2574438 4.226760 0.02138597 0.3647961 3.798588 3.243232 4.411147
8 0.2354753 4.227439 0.02758202 0.5159806 4.146887 3.467366 4.939591
9 0.2512287 5.066621 0.03813687 0.8212202 4.638575 3.750960 5.641951
10 0.2830375 6.905185 0.05862062 1.5639190 5.606438 4.260455 6.477177
In this case, the bx estimates vary, but not strongly and with no evident pattern – so I would be fairly confident in the reliability of the methods (which in this case both suggest a positive and increasing causal effect – evidenced by the positive and increasing values of by/bx).
Estimates from the doubly-ranked method can be fed into the fractional polynomial or piecewise linear plotting functions similarly to those from the residual method:
with(summ_data2$summary, piecewise_summ_mr(by, bx, byse, bxse, xmean, xmin,xmax,
ci="bootstrap_se",
nboot=1000,
fig=TRUE,
family="gaussian",
ci_fig="ribbon"))
We hope this tutorial helps introduce you to the doubly-ranked method. More details on the software can be found here: https://github.com/amymariemason/SUMnlmr.
- Details
- Written by: Steve Burgess
When performing a Mendelian randomization analysis based on a single gene region, it is possible to include only a single variant in the analysis. However, if multiple variants explain independent variance in the exposure, then the analysis will be more efficient using all those variants - even if they are partially correlated. With summarized data, this can be achieved using the inverse-variance weighted (IVW) method, by incorporating a correlation matrix in the analysis.
For this, it’s important that the correlations are in the right direction – what is needed are signed correlations, not squared correlations (r, not r^2). The signs of the correlations must correspond to the same effect alleles as the genetic associations. It's best to estimate such a matrix in a large sample with similar ethnic background to the dataset from which the summarized data are taken (in particular, the genetic associations with the outcome). If this isn't possible, then the ld_matrix in the TwoSampleMR package can help. For example:
rho = ld_matrix(c("rs7529229", "rs4845371", "rs12740969"))
rho
> rs7529229_C_T rs4845371_T_C rs12740969_T_G
> rs7529229_C_T 1.000000 -0.687196 -0.571108
> rs4845371_T_C -0.687196 1.000000 0.155994
> rs12740969_T_G -0.571108 0.155994 1.000000
We see that the effect alleles are T for rs7529229, C for rs4845371, and G for rs12740969. If our summarized data instead used the T allele for rs4845371, we can flip the relevant elements of rho in the second row and column:
flip = c(1, -1, 1)
rho.new = rho*flip%o%flip
One question is this: suppose there are multiple genetic variants in a given gene region that are all potential instruments. How to decide how many variants to include in a Mendelian randomization analysis? Including too few variants may result in inefficiency. But including too many variants in the analysis may result in unstable estimates. The reason is that including highly correlated variants can result in numerical instabilities when inverting the correlation matrix - small changes in the correlation matrix can lead to large changes in the MR estimate. The same happens if you include 3 or more variants that aren’t close to 100% pairwise correlated, but they all predict each other (the technical term is “linearly dependence” or “multicollinearity”).
While we have developed methods for highly correlated variants - for example, here Mendelian randomization with fine‐mapped genetic data: Choosing from large numbers of correlated instrumental variables (nih.gov) we suggest using principal components analysis (PCA) to summarize the genetic variants - these methods are not foolproof when the correlation matrix is imprecisely estimated or does not fit the data well (perhaps it is estimated in a slightly different population). This can result in an overly precise estimate. A similar method (but a little more robust to weak instruments) can be found here: [2005.01765] Inference with many correlated weak instruments and summary statistics (arxiv.org)
Practical advice is this: first, start with aggressive pruning – would suggest a threshold of r^2<0.1, and account for correlations in the analysis. By increasing the pruning threshold (to r^2<0.2, r^2<0.3, etc) and including additional variants, you can potentially get an estimate that is slightly more precise that this, but if the standard error reduces sharply (say it decreases by a factor of 3), then I wouldn’t trust the estimate, and instead would suggest reporting the estimate with more aggressive pruning. Similar when using the dimension reduction methods - if the MR estimate gets a bit more precise, then it’s probably reliable, but if it’s substantially more precise, then I’d be concerned.
From experience, pruning at r^2 < 0.3 is generally safe, and r^2 < 0.4 is usually okay – but I’ve seen problems at this level in a couple of examples. If at all possible, I’d recommend trying to get correlation estimates in as large a sample size as possible (but still relevant to the dataset under analysis!) - and be suspicious if including additional highly correlated variants to an analysis substantially reduces standard errors!
- Details
- Written by: Steve Burgess
One question I’ve been asked a large number of times is how to perform a power calculation in Mendelian randomization for a binary exposure. We wrote a longer paper on the difficulties of MR with a binary exposure previously (Mendelian randomization with a binary exposure variable: interpretation and presentation of causal estimates), but we removed details on power calculation due to limited word count.
The short answer is that it's not simple, and potentially not possible unless you have additional data. If the gene—exposure associations are estimated on the odds ratio scale, it makes a big difference to power if the exposure is rare (e.g. for an odds ratio of 1.2, the genetic variant would increase exposure prevalence from 0.1% to 0.12% in absolute terms) or the exposure is common (e.g. the genetic variant increases exposure prevalence from 10% to 12%). Power depends on the absolute magnitude of the increase in the exposure prevalence, not the relative increase on the odds ratio scale. So if you have no way of estimating genetic associations with the exposure on an absolute scale (such as linear regression of the exposure on the genetic variant), then you can't calculate power.
If you can get the IV—exposure associations on an absolute scale (e.g. the genetic variant increases the prevalence of the outcome by 2% - say from 10% to 12%), then you can calculate power. But power calculations are still not simple as power is normally given for a proposed effect size per 1-SD unit increase in the exposure, and a 1-SD change for a binary exposure may be a little difficult to conceptualize. Instead, we can consider the proposed per 1 unit increase in the exposure (from 0 to 1). In this case, you can use standard tools for power calculation (such as Online sample size and power calculator for Mendelian randomization with a binary outcome), but instead of the R^2 measure, you need a different value. This is obtained by taking the beta-coefficient from linear regression of the exposure on the genetic variant (call this number beta). For a biallelic SNP, instead of the R^2 statistic, you want beta^2*2*MAF*(1-MAF).
So if you have a genetic variant that is a SNP with a minor allele frequency of 0.3 that increases the prevalence of the exposure by 10%, then your replacement “R^2 value” is 0.1^2*2*0.3*0.7 = 0.0042. Power to detect a 0.3 SD increase in the outcome per 1 unit increase in the exposure in a sample size of 10,000 would be around 50%, as shown in the screenshot below:
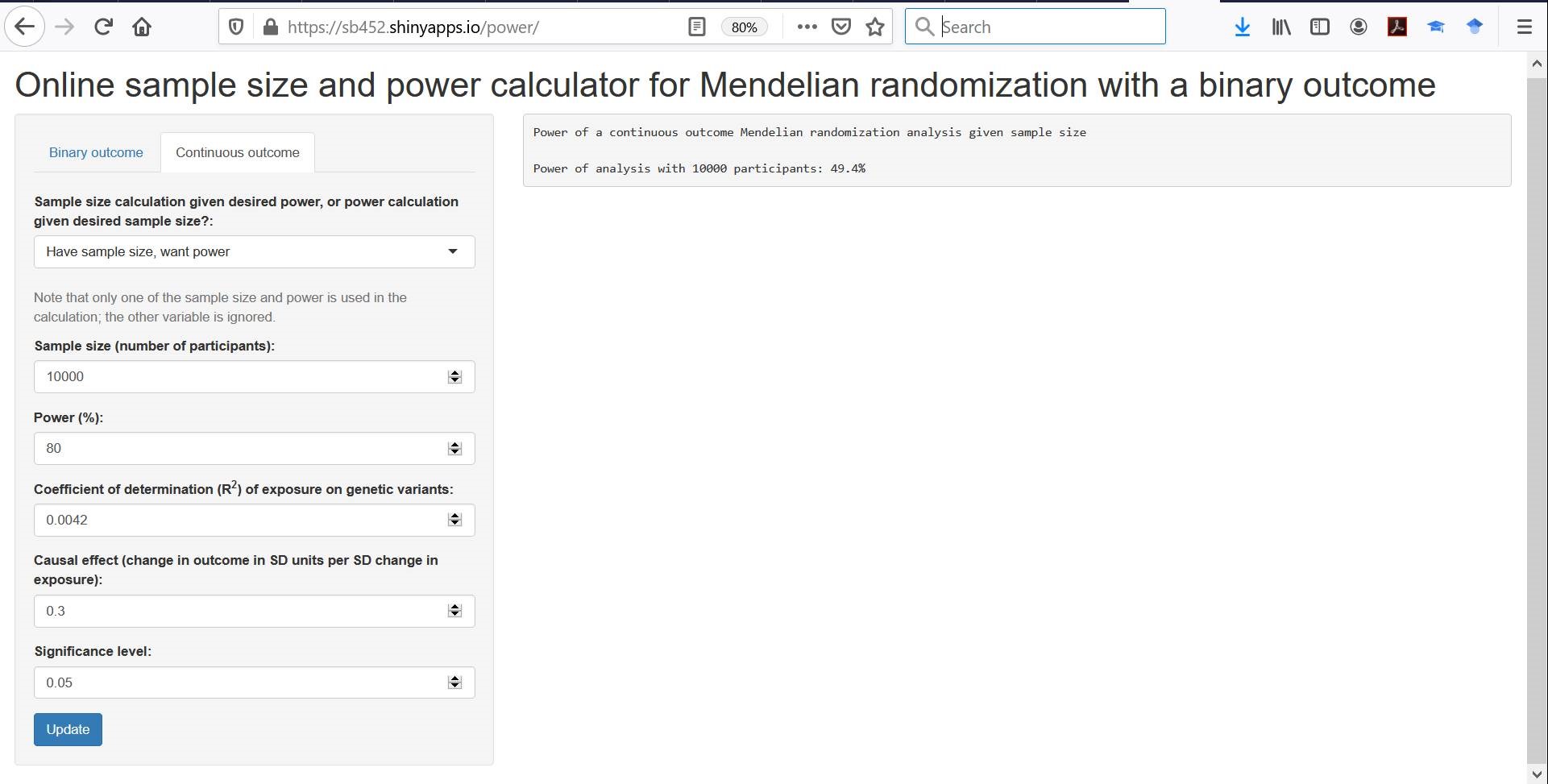
A quick simulation exercise demonstrates that this value is fairly accurate:
set.seed(496)
times = 1000
parts = 10000
pval = NULL
for (j in 1:times) {
g = rbinom(parts, 2, 0.3)
x = rbinom(parts, 1, 0.2+0.1*g)
y = rnorm(parts, 0.3*x)
pval[j] = summary(lm(y~g))$coef[2,4]
}
sum(pval<0.05)
> [1] 492
Hope that is helpful!
- Details
- Written by: Steve Burgess
Given the global situation with reduced travel and face-to-face content, it is not possible to plan an in-person course in the foreseeable future. So, the November 2020 version of the Mendelian randomization course will be delivered remotely via an online learning platform. We are currently working through details of how this will work - please be patient and bear with us while we work through the details!
UPDATE: Course is now full! We are hoping to repeat in Spring 2021.